
Locations
Yesterday, on 1st August 2024, the AI Act ("AIA") entered into force. Designed as a product regulation, it regulates AI systems and general-purpose AI models. The AI Act has a major impact on the health care sector, where AI systems are already widely used, e.g., to diagnose diseases, as a clinical decision support, to classify and analyse health data, to optimise medical therapy and treatment, but also in telemonitoring tools, healthcare robots, patient's wearables or medical chatbots. The comprehensive obligations of the AI Act add on to an already highly regulated sector and a lengthy procedure for placing products on the market.
Call to action:
Stakeholders providing, using, distributing or importing AI systems in the health care sector should act now to ensure timely compliance. This includes assessing the scope of applicability of the AI Act, conducting a detailed gap analysis with regard to the obligations set out in the MDR and IVDR, working closely with notified bodies and following upcoming guidance and technical standards of the EU Commission.
We are happy to help you navigate the comprehensive obligations and evaluate the options that best suit your business needs.
In the following, we outline a few aspects you need to consider.
Scope of application and definition of AI systems
The obligations for AI systems apply to all AI systems placed on the market or put into service in the EU irrespectively of the establishment in the EU or a third country. Moreover, it is applicable when the output produced by the AI system is used in the EU.
In line with the OECD definition, the AI Act defines AI systems as:
"a machine-based system that is designed to operate with varying levels of autonomy and that may exhibit adaptiveness after deployment, and that, for explicit or implicit objectives, infers, from the input it receives, how to generate outputs such as predictions, content, recommendations, or decisions that can influence physical or virtual environments"
Only a few cases do not fall within the scope of the AI Act. This includes AI systems that are specifically developed and put into service for the sole purpose of scientific research and development (R&D), when the operator only planned to conduct research, testing or development activities or the AI system is or will be offered under a free and open-source license. The distinction may be difficult, particularly with regard to R&D and the conduct of clinical investigations/performance studies (see also below).
Risk-based approach
The complex regulatory structure of the AI Act follows a risk-based approach. AI systems are therefore categorised as unacceptable risk, high-risk, limited or transparency risk and minimal risk.
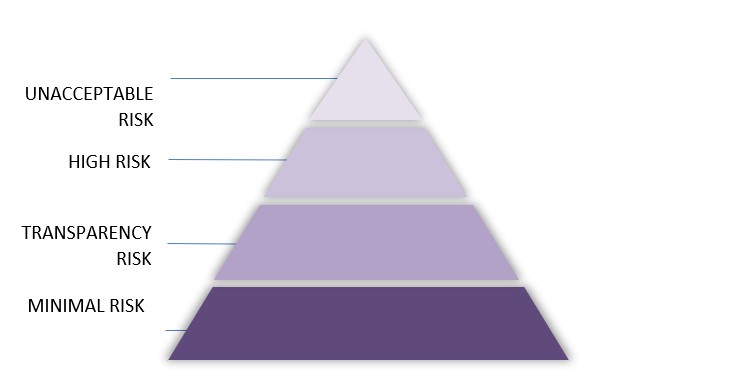
The AI Act provides for comprehensive obligations, particularly with regard to high risk-AI systems. This is of great relevance to the health care and MedTech sector, as most of the medical devices incorporating artificial intelligence are classified as high-risk AI systems. According to Art. 6 (1) AIA, AI systems shall be considered high-risk where:
- the AI system is intended to be used as a safety component of a product, or is a product itself,
- the product is covered by the Union harmonisation legislation, e.g., the Medical Device Regulation (MDR) or the In-vitro-medical device regulation (IVDR), and
- the safety component or the product itself is required to undergo a third-party conformity assessment, with a view to the placing on the market or putting into service of that product pursuant to the Union harmonisation legislation.
However, the AI Act provides for a "filter system" according to which – by derogation of Art. 6 (1) AIA – AI systems that
- perform narrow procedural tasks,
- improve the result of a previously completed human activity,
- detect decision-making patterns or deviations from prior decision-making patterns not meant to replace or influence the previously completed human assessment, without proper human review, or
- perform a preparatory task to an assessment
are not subject to the obligations for high-risk AI systems. Such systems do only have to comply with the documentation and registration requirements. Therefore, it is important to analyse in detail whether the AI system falls within the comprehensive obligations of high-risk AI systems or is covered by the filter system.
Actors and obligations
The AI Act addresses different operators to comply with the obligations:
- providers (a natural or legal person, public authority, agency or other body that develops an AI system or a general-purpose AI model or that has an AI system or a general-purpose AI model developed and places it on the market or puts the AI system into service under its own name or trademark, whether for payment or free of charge; e.g., medical device manufacturers)
- deployers (a natural or legal person, public authority, agency or other body using an AI system under its authority except where the AI system is used in the course of a personal non-professional activity; e.g., hospitals, practitioners, users)
- importers (e.g., a natural or legal person placing an AI system on the market that bears the name or trademark of a natural or legal person established outside the Union)
- distributors (e.g., a natural or legal person, other than the provider or importer, that makes an AI system available on the EU market)
The provider is subject to most obligations. This includes obligations on registration, data and data governance, technical documentation, record keeping, human oversight, accuracy, robustness and cybersecurity, quality management system, risk management system, post-market monitoring, conformity assessment, EU declaration of conformity and CE marking.
Particular attention should also be paid to cases where you may be considered a provider even though you (only) use and deploy the AI system. This is the case when
- you put your name or trademark on the high-risk AI system already placed on the market or put into service,
- you make substantial modification to a high-risk AI system that has already been placed on the market or put into service in such a way that it remains a high-risk AI system,
- you modify the intended purpose of an AI system, which has not been classified as high-risk and has already been placed on the market or put into service in such a way that the AI system concerned becomes a high-risk AI system.
Whether your activity falls into one of these cases, should be carefully examined, as you will have to fulfil considerably more extensive obligations. We are happy to advise you on the best options for your business needs.
Specific challenges for the health care sector
We can see from our experience that there are some very specific challenges for the health care sector which we elaborate on in the following paragraphs.
Interplay of the AI Act and the MDR and IVDR
The MDR and the IVDR already regulate medical devices and in vitro diagnostic medical devices qualifying as artificial intelligence-based software. These regulations also provide similar obligations to the AI Act, such as those relating to technical documentation, risk management and conformity assessment. However, the AI Act envisages that there shall be no duplications (see with regard to the conformity assessment Recital 124). Consequently, it is required to carefully analyse which requirements are to be met additionally to the MDR and IVDR.
Continuous learning AI systems
It is also challenging to assess the conformity of continuously learning AI systems. In particular, it should be carefully evaluated when a continuously learning AI systems has to undertake a new conformity assessment. The AI Act draws the line where the AI system is subject of a substantial modification as long as its performance has not been pre-determined by the provider at the moment of the initial conformity assessment (Art. 43 (4) AIA).
Conducting clinical investigations/performance studies
Clinical investigations and performance studies are crucial for the development of medical devices. Therefore, a detailed analysis is needed when such investigations and performance studies fall under the AI Act as it is to be considered as "putting into service" or whether the foreseen practice falls outside of the applicability of the AI Act as it is considered to be R&D (see above).
Notified bodies
Notified bodies that are competent to assess medical devices can also be designated as notified bodies in accordance with the AI Act. This enables setting up a joint CE marking, conformity assessment and technical documentation claiming compliance with the MDR, IVDR and AIA. However, not every notified body may have the necessary personnel and capacities. It is therefore crucial to stay in close contact with the notified bodies and, if necessary, to take timely steps to switch to another body that is qualified with respect to AI systems.
Placing on the market
The comprehensive obligations of the AI Act expand the already highly regulated health care and MedTech sector and the lengthy process of placing products on the health care market. This will have an impact on the resources (time, budget, etc.) allocated to the development and deployment of medical devices. It is crucial to be aware of the fact that specific obligations should be implemented in the development process. This applies in particular to the obligations relating to data and data governance as it might be difficult or even impossible to subsequently adapt the underlying data and data governance subsequently if the notified body ultimately considers that compliance has not been achieved.
Risks and penalties
When not complying with the obligations of the AI Act high fines may be imposed:
- An AI system that falls within the category of unacceptable risk and may therefore not be placed on the market or put into service may be subject to a fine up to EUR 35,000,000 or 7% of the total worldwide annual turnover for the preceding financial year.
- Considering a high-risk AI system that does not comply with the corresponding obligations, a fine up to EUR 15,000,000 or 3% of the total worldwide annual turnover for the preceding financial year may be imposed.
We are happy to help you navigate the comprehensive obligations and evaluate the options that best suit your business needs.
Supporting author:
Lea Köttering is a Trainee in the Tech & Data Team at the Fieldfisher office in Hamburg. She focuses on data protection law and artificial intelligence, with a particular interest in applications and developments in the life sciences sector.